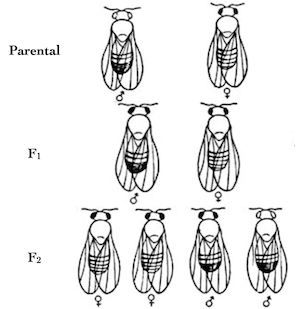
I put together a rambling video about the final project in my genetics class, and about the responsibility of modern geneticists to deal with the terrible bad ideas of the past — eugenics. I give you a few examples of bad genetics, one relatively benign, and another actively evil (as you might guess, the evil example is Donald Trump.)
The gentler example is Fairfax Cryobank, which provides a good, useful, and even necessary service, sperm storage. I’ve visited their St Cloud branch, not as a client, but leading a field trip for a class on modern reproductive technologies, and they seem like good people with a lot of dewars. You can browse their catalog of sperm donors, and it’s a real trip. It’s more like reading the submissions to a dating site…a dating site where you’ll never meet the person whose profile you’re reading, but if you’re lucky and spend a few thousand dollars, you might get a frozen vial of sperm in the mail.
Here, for example, is one profile among many.
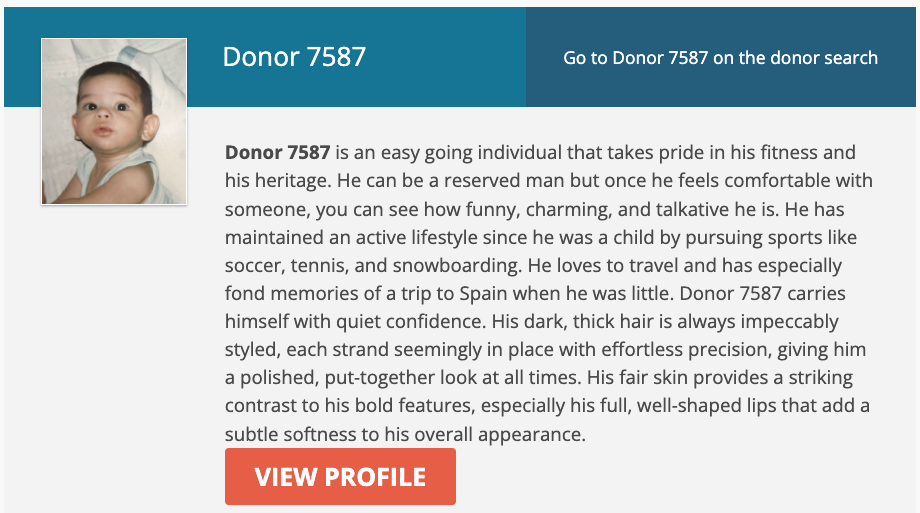
Donor 7587 is an easy going individual that takes pride in his fitness and his heritage. He can be a reserved man but once he feels comfortable with someone, you can see how funny, charming, and talkative he is. He has maintained an active lifestyle since he was a child by pursuing sports like soccer, tennis, and snowboarding. He loves to travel and has especially fond memories of a trip to Spain when he was little. Donor 7587 carries himself with quiet confidence. His dark, thick hair is always impeccably styled, each strand seemingly in place with effortless precision, giving him a polished, put-together look at all times. His fair skin provides a striking contrast to his bold features, especially his full, well-shaped lips that add a subtle softness to his overall appearance.
They’ve all got cute little baby pictures, since you won’t meet the adult. This is all for the benefit of clients, who will pick a vial of sperm based on vibes, but almost everything in that description is not heritable. You won’t get a vial filled with “funny, charming, and talkative,” because those are things that family, friends, and experience will generate. My objection is that it perpetuates the myth of simple inheritance of traits for everything, and misleads the client. But all of reproduction is a misleading game, as far as the traits of your child are concerned.
I would recommend adding a more appropriate button to the website: a “RANDOM CHOICE” button. Click it, they’ll send your doctor a completely random arbitrary vial from their vast collection. You’ll be surprised! But no more surprised than if you carefully choose the father of your child based entirely on a profile on a website.