I’ve been complaining about the cancer nonsense peddled by Paul Davies and Charles Lineweaver for years. I imagine nothing will stop them. They’ve got this weird idea that cancers are atavisms — that what they are is a reactivation of an ancestral program that was constructed a billion years ago by free-living single-celled organisms, that was then shackled and constrained by the evolution of multicellularity, but which can then re-emerge when the old program is liberated by mutations in the multicellularity genes that are its jailers. Cancers are your protistan ancestors, yearning to break free.
It’s nonsense. Davies and Lineweaver are physicists with no comprehension of how evolution works. There are no ‘genetic programs’ that can linger for a billion years in a suppressed state to functionally rebound with the accidental removal of a metazoan control program. When you add that Davies justifies his model by invoking Haeckelian recapitulation, a theory that’s been dead and wrong for over a century, you’ve got a recipe for raging crackpotterism from a sober, respected physicist.
Yet they still get published. They still manage to sucker in scientists who ought to know better. They’ve published again in PLoSOne, in a paper titled “Ancient genes establish stress-induced mutation as a hallmark of cancer”. Here’s the abstract.
Cancer is sometimes depicted as a reversion to single cell behavior in cells adapted to live in a multicellular assembly. If this is the case, one would expect that mutation in cancer disrupts functional mechanisms that suppress cell-level traits detrimental to multicellularity. Such mechanisms should have evolved with or after the emergence of multicellularity. This leads to two related, but distinct hypotheses: 1) Somatic mutations in cancer will occur in genes that are younger than the emergence of multicellularity (1000 million years [MY]); and 2) genes that are frequently mutated in cancer and whose mutations are functionally important for the emergence of the cancer phenotype evolved within the past 1000 million years, and thus would exhibit an age distribution that is skewed to younger genes. In order to investigate these hypotheses we estimated the evolutionary ages of all human genes and then studied the probability of mutation and their biological function in relation to their age and genomic location for both normal germline and cancer contexts. We observed that under a model of uniform random mutation across the genome, controlled for gene size, genes less than 500 MY were more frequently mutated in both cases. Paradoxically, causal genes, defined in the COSMIC Cancer Gene Census, were depleted in this age group. When we used functional enrichment analysis to explain this unexpected result we discovered that COSMIC genes with recessive disease phenotypes were enriched for DNA repair and cell cycle control. The non-mutated genes in these pathways are orthologous to those underlying stress-induced mutation in bacteria, which results in the clustering of single nucleotide variations. COSMIC genes were less common in regions where the probability of observing mutational clusters is high, although they are approximately 2-fold more likely to harbor mutational clusters compared to other human genes. Our results suggest this ancient mutational response to stress that evolved among prokaryotes was co-opted to maintain diversity in the germline and immune system, while the original phenotype is restored in cancer. Reversion to a stress-induced mutational response is a hallmark of cancer that allows for effectively searching “protected” genome space where genes causally implicated in cancer are located and underlies the high adaptive potential and concomitant therapeutic resistance that is characteristic of cancer.
A translation:
-
Some have said that cancer is a reversion to the single-celled state. The “some” just happens to be us.
-
We naively predict that the genes involved in a disease of multicellularity, cancer, would be genes that have evolved after the emergence of multicellularity. We helpfully defined a colossal window of one billion years to assay.
-
We compared the distribution of cancer-causing mutations to a uniform, random distribution of mutations. Surprise: some mutations cause cancer, others don’t, so it doesn’t fit a random distribution. There are lots of genes that are less than 500 million years old that are implicated in cancer.
-
But wait! When we looked in a database of cancer-causing gene mutations, COSMIC, we find that the database is enriched for old genes. This is contrary to our hypothesis, therefore we need to find a new rationalization, rather than rejecting our hypothesis.
-
The genes that are commonly broken in cancer are involved in DNA repair and cell cycle control, which are truly primitive, ancient functions — prokaryotes have genes for this. What possible “ancient program” could be reactivated to fit our story?
-
I know! Stress-induced hypermutation! Cancer cells are invoking multi-billion year old processes; it can’t be that breaking genes produces a loss of specificity and efficiency in repair, they are switching on genes to cause mutations. They’re doing it on purpose! Yeah, that’s the ticket!
It’s a truly awful mess of a paper, in which the authors juggle lots of data to make it fit their preconceptions, and in which apparently no result can possibly cause them to reject their hypothesis. And their conclusions are strange.
Here we present evidence demonstrating that cancer manifests as an atavistic recapitulation of pre-metazoan mechanisms of stress-induced mutation in somatic cells, explaining its capacity to evolve resistance to therapy. The mechanistic roots of this behavior are retained over evolutionary time scales because they are critical to the successful function of the germline and immune system. In addition to generating base-line diversity in both the innate and adaptive immune system, normal germline mutational patterns maintain diversity in recently evolved gene families governing functions such as toxin detection and detoxification. In cancer the controlled restriction of this phenomenon to the germline and immune system is disrupted, allowing somatic cells to effectively search ancient genome space for solutions to the stress-induced pressures they are experiencing. We propose stressed-induced mutation as a hallmark of cancer reflected by genomic instability.
There is a phenomenon in the immune system where, for instance, the immunoglobulin genes are prone to greater mutation rates during cell proliferation — it is a way to increase the diversity of antibody types. So yes, there are mechanisms to increase the rate of errors. What Davies and Lineweaver are proposing is that cancer cells are actively and purposefully switching on these mutation-generating processes to “search ancient genome space”, whatever the hell that is and however the hell inducing a greater frequency of random mutations would find and restore these imaginary “ancient genes”. Note also that they’re babbling about “atavistic recapitulation of pre-metazoan mechanisms” while discussing properties of the adaptive immune system. This makes no sense.
Genomic instability is one of the hallmarks of cancer. It’s not necessarily a planned sort of thing. If you disrupt DNA repair mechanisms with a mutation, you’ll get more mutations in other genes. If you mutate the gatekeeping proteins that act to ensure that cell division does not proceed if you have gross chromosomal errors, you will have more gross chromosomal errors. The fact that populations of cancer cells routinely accumulate new mutations as they progress is not an observation that supports the idea that cancers are a reversion to an ancestral healthy, single-celled state.
If you need to cleanse your palate after that dog’s breakfast of bad science, here’s a much more interesting paper on cancer: Carbon dating cancer: defining the chronology of metastatic progression in colorectal cancer. The investigators took advantage of a tragic series of events in a cancer patient: an initial biopsy initiated needle track seeding. That is, a few cells trickled out of the biopsy needle, which acted as the mechanism of metastasis, and started new tumors, so they knew precisely when these tumors were initiated. This allowed them to use standard phylogenetic methods on sequences from samples of the cancer as it progressed, and put together a history of mutations for the diversifying cells of the cancer.
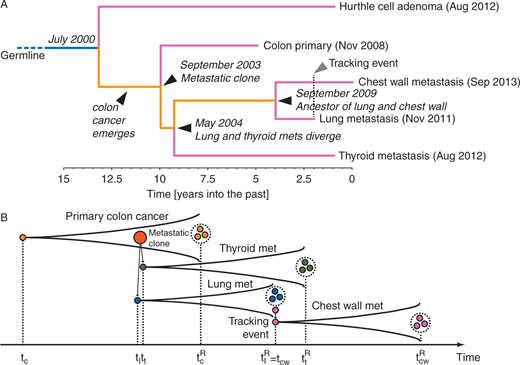
Chronology of the patient’s CRC evolution. (A) Whole-genome sequencing of multiple lesions from the patient’s malignancy allowed phylogenetic reconstruction of the tumour tree. In the phylogenetic tree, dates within brackets indicate the time of clinical diagnosis whereas dates in italic highlight the estimated times of the different lesions. (B) Illustrative cartoon of the patient disease progression and samples taken. At time tc the first colorectal cancer cell arose, giving rise to the primary tumour. At time tl the first metastatic clone emerged, giving rise to the lung metastasis, quickly followed by a second metastasis to the thyroid emerging at time tt . At time tcR the sample from the primary tumour was collected (resection) and analysed. During the lung biopsy, the needle tract seeding event spread cancer cells in the chest wall at time tcw. A few weeks later, at time tcR the lung metastasis was resected and profiled. Finally at time tcwR the chest wall metastasis was also sampled.
I know this excerpt from the paper is rather dense, but all you need to take away from it is that the different branches of the cancer pedigree have some common mutations, and also a constellation of unique mutations. Look, TP53, often called the “guardian of the genome”, is completely taken out with a missense mutation, and whole chromosomes show LOH (loss of heterozygosity) — that is, the cancer cells have lost entire copies chromosome 5, and in one subset, chromosomes 2, 9, and 22. This kind of wholesale chaos would not have been an adaptive response by a protistan ancestor.
Targeted sequencing of 409 cancer-related genes in both the primary CRC and all metastatic sites (lung, thyroid, chest wall and urinary tract) of our case revealed the presence of clonal non-sense mutations in APC (Gln1367*), as well as missense mutations in CTNNB1 (Leu156Gln), KRAS (Gly12Asp) and TP53 (Ser215Ile). The same mutations were not detected in the normal tissue or in the Hurthle adenoma. Two genes included in our panel showed discordance between primary and metastatic cancer: ADAMTS20, a metalloproteinase involved with cancer invasion and migration, and AKAP9 an A-kinase anchor protein which binds to the regulatory subunit of protein kinase A. The ADAMTS20 missense mutation Arg1885Thr was observed in the primary cancer but was not detected in either the lung, thyroid or chest wall metastases. Conversely, the AKAP9 missense mutation Ala3077Pro was found in all the metastatic sites but was not detected in the primary cancer. All these mutations were also found using WGS [Whole Genome Sequencing], furthermore ADAMTS20 and AKAP9 were validated by Sanger Sequencing. All lesions were microsatellite stable (MSS). Copy number analysis based on WGS data revealed a relative low level of chromosomal instability (CIN), with LOH of chromosome (chr) 5, gain of chr7, and a focal amplification on chr13q12.2-12.3, encompassing the VEGFR1 and CDX2 genes. These aberrations were clonal in all the CRC lesions, whereas the Hurthle adenoma showed a distinct profile characterized only by loss of chr2, chr9, and chr22. Primary tumour and metastatic sites displayed the same dominant, age related, mutational signature 1 which has previously observed in CRC as well as other cancer types.
Now that’s interesting stuff. Unfortunately, one of the lessons learned from analysis of other cancers is that every cancer is different — there are common modalities, such as the loss of certain checkpoint proteins, but there are multiple ways to achieve certain outcomes, and they don’t have to appear in any particular order. It definitely looks far less like a reactivation of a specific ancestral state and more like a derangement of regulatory functions.
Davies’ approach is kind of silly, too, a test designed to give whatever results the researchers want, with a set of observations that were basically a given with their parameters. For example, a lot of cancers mess up cell signaling to trigger uncontrolled growth. The signaling molecules are often factors like receptor tyrosine kinases (RTKs), which are only found in multicellular animals and one group of protists. So yes, it’s inevitable that you’ll find a lot of “young” (less than a billion years old) genes among your cancer-causing candidates. That observation does not support the idea of cancers being atavisms.

Davies and Lineweaver are physicists
Par for the course. You must have noticed that most people with “revolutionary” (i.e totally off-the-wall) ideas have no conceivable connection with the discipline they are talking about.
I tend to look at the popular psych books or the new revolution ally diet books on the library’s new books shelf or in the book store. None of the authors have any relevant training or experience.
The Food Chick seems to have some background in computer science, the author of Wheatbelly is a cardiologist, and so on.
Blast, I cannot think of any of the popular psych books, though I suppose maybe Dale Carnegie might be included. I must not be keeping up with my reading.
Of course, once in a long while, someone who really is an expert will write a book, like Daniel Kahneman’s Thinking fast and slow but they are as rare as hen’s teeth. And even Kahneman’s book is not a flashy cure your shyness and improve your sex life in 10 days type of book
More typically it is like having Ben Carson write a book on Egyptology with special emphasis on the pyramids.
Davies speaking of another “discovery”
“I had the advantage of being unencumbered by knowledge. I dropped chemistry at the age of 16, and all I knew about arsenic came from Agatha Christie novels.”
Glad you’ve got it covered. I tried to keep up, but got snowed under early on. I was buried in something anyway.
I’ve come across enough people spouting nonsense outside their sphere of competence to know it’s as common as muck. The interesting question is: why do ASU and the NCI continue to support this nonsense? Don’t they listen to their biologists?
Davies is very well-funded, getting money from the Templeton foundation as one of their very serious scholars.
jkrideau:
Here’s an extract from a February post at RetractionWatch that you might find of interest.
Drat, I befouled the mark-up. That last sentence is mine, not a quotation. Sorry.
Is this the same Paul Davies who’s been known for years for his theistic apologetics, linking a vague Mind of God vaguely with modern physics? The scientist regularly touted as a smart person who rejects atheism for smart reasons? If so, then I’m not surprised he’s getting weird with evolution. He’s coming at it with some prior baggage.
Most physicists do not regard Davies as “a sober, respected physicist”. Many consider him kooky (if not fully crack-potted) in their own field, of physics. He tends to invoke God a lot and adheres to loony interpretations of ‘fine-tuning’ arguments and anthropic notions as ‘evidence’ of an intelligent design behind it all that specifically prepared the entire universe (and us) for our personal benefit.
PZ: I strongly disagree.
It’s hard to call it a bandwagon when there’s only 2 people. I assert that they are back on the hobbyhorse.
I hope you feel appropriately abashed.
Seedy
You are getting right fuckin’ awesome at the Lawyerspeak.
I may hire you, someday soon.
Ye gods.
Well, if those ancient genes are functional originally and then become dysfunctional through mutation, then somatic cells were able to “search ancient genome space” more effectively before they become cancerous.
And if exposure to mutagens is a stress-inducing pressure, how exactly is becoming an invasive tumour line a “solution” to anything? You might as well say breaking your femur is a stress-induced solution to the problem of being in a car crash.
And, why, if cancer is an atavistic reversion to a unicellular state, is one of the strong predictors of cancer progression the development of complex multicellular tissue structures such as arteries that feed the tumour?
Trump’s Syndrome?
Is this statement the right way ’round? Do cancers cause messed up signaling, or does messed up signalling cause cancer?