Thirty, almost forty, years ago when zebrafish were an up and coming model system and very few labs were working on them, we were used to going to conferences and reciting the zebrafish litany, a list of attributes that justified us working on such an oddball animal: we’d explain, for instance, that it was prolific, fast developing, and optically transparent, so we could see right into the nervous system in the living embryo. And you know, not once have I ever been asked the really simple and obvious question: if it’s transparent, then how do we see anything inside it?
I know. It takes a moment to step outside the assumptions (we say we see stuff, after all) and think about what transparency actually means. Of course, I always have an answer prepared: “With differential interference contrast microscopy, Nomarski optics specifically.” A polysyllabic phrase is always an effective way to shut down the rubes, isn’t it?
But I can also explain it in simple English, especially if I can flail about on a chalkboard at the same time. So let’s try.
We’ll start with the very minimal basics. Light is a wave, and the properties of that wave are what our eyes see. For instance, one property of a wave is amplitude: in the two light rays below, the lower one has a larger amplitude, so we’d see it as having a greater intensity, or being brighter, than the top one.

Another property is wavelength or frequency. Shorter wavelengths (higher frequencies) have have more energy than longer wavelengths (lower frequencies), and we see these differences as color. In the cartoon below, the longer wavelength of light would be seen as red, while the shorter one is blue.
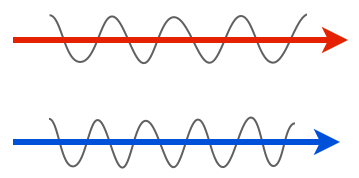
Those are the characteristics of light that our eyes can detect, so when I say that zebrafish embryos are transparent, I mean that light passing through them does not exhibit a significant reduction in amplitude or a change in wavelength — it does not get significantly dimmer nor does it change color.
But there are other important properties of light. One is phase. Notice how in the previous picture of the amplitude, the peaks in one line up with the peaks in the other, and the troughs line up with the troughs? We’d say those two rays of light are in phase. They don’t have to be, though. For instance, here are two rays of light that are completely out of phase, with the peaks of one lining up with the troughs of the other.
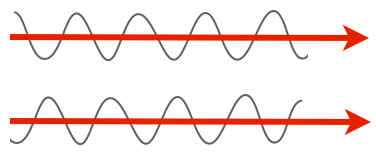
Here’s the thing, though: our eyes can’t discriminate the phase of light. Both of those beams have the same amplitude and the same wavelength, so they’d look completely identical to us. Which is a shame, because lots of transparent things shift light phases, and especially of interest, lots of biological materials shift the phase. All you need to do is have a material with a different refractive index and you’ll get a phase shift.
For example, most of a cell is water, and it’s surrounded by a membrane that is mostly oil/fats. These have different refractive indices. Shine two beams of light on a transparent cell, one that passes through the center (mostly water) and one that passes tangentially through the membrane (lots of oil), and you end up with two beams of light that now have different phases. Which we can’t see, darn it. My zebrafish embryos are full of membranes in complex arrangements, so while light passes through them with amplitude and wavelength (intensity and color) relatively unaffected, the phases of light are all scrambled and cockeyed in interesting ways full of information. We just have to have a way to detect phase and we could see lots of phenomena in the embryo.
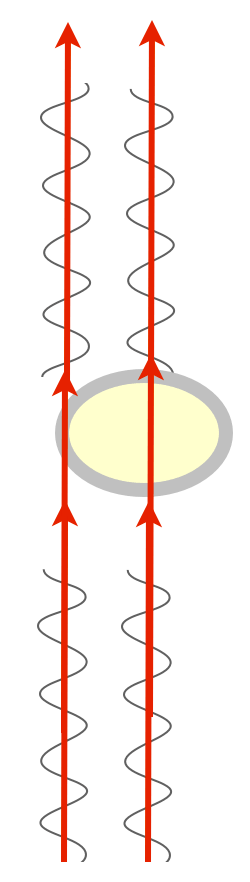
There is a way to visualize the relative phase of two beams of light, though. If you could just bring them together, combine them, they could interfere with one another. If the two beams are in phase, peaks lining up with peaks, they’d sum up and you’d see a greater amplitude — the combined beam would be brighter. This is called constructive interference. If they’re out of phase, peaks lining up with troughs, they’d cancel each other out, they’d destructively interfere, and you’d get a dimmer beam.
We have this really clever way of combining two beams of light: they’re called prisms and lenses, which bend light. All we have to do is shine parallel beams of light through our specimen, and then use prisms to bend one and bring it into alignment with the other, and we’d get interference patterns that change the intensity of the light, and we’d be able to detect phase changes!
There is a tricky bit to this, though. This is a way of comparing the phase of two rays of light; what are we going to compare? One technique is to illuminate with ring of light, and use light that hasn’t passed through your specimen as a reference beam, and compare that to light perturbed by your specimen, the specimen beam. This is the technique used in standard phase contrast microscopy. It has some drawbacks I won’t get into in any detail; you get halos that reduce the resolution of your image. We don’t like halos. It’s kind of like the lens flares that cheesy sci-fi cinematographers sprinkle into their movies.
Differential interference contrast uses a different method to generate a reference beam. We illuminate with two beams of light that have different polarity and that are offset slightly (by a fraction of a micrometer) from one another. Polarity is another of those properties of light that our eyes can’t really detect; Dr Dryskull has an excellent post on polarity, but simply put, those waves of light aren’t all necessarily vibrating in the same plane — they can be wiggling in all sorts of directions. We use a Wollaston prism, a piece of crystal that splits light by its polarity, so that we have light vibrating in one plane, and another beam vibrating at 90° to the first, both shining through the specimen in parallel. Their phase will be affected in different ways because each will be passing through a very slightly different part of the specimen. Then they are recombined by passing through a second Wollaston prism, allowing them to interfere with one another, to produce interference patterns.
Does this diagram help? Basically you have all kinds of glass in the condenser and above the lens that jigger the heck out of light to split, isolate, and recombine the beams into visible patterns of constructive and destructive interference, allowing you to visualize shifts in phase, which represent changes in the refractive index, of cells and tissues.
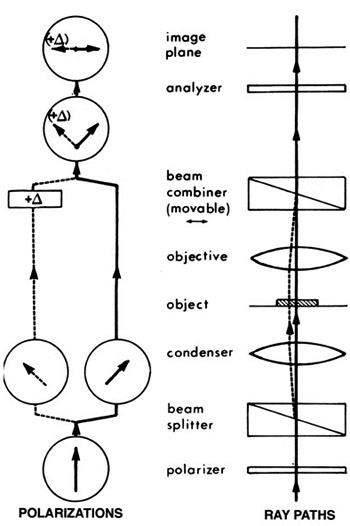
More simply put, this technique compares the phase shift of each point on the specimen to a point 0.2 microns to the right (or whatever; we have knobs that let us shift the relative orientation). If you’re looking at a uniform field, this comparison yields the same intensity everywhere; but at regions in transition, for instance where phase is changing rapidly at a membrane boundary, you get bright and dark edge enhancement. This turns a bland, flat field of cells into a picture of clustered basketballs with a kind of lovely shadowing effect. Like so:

It can also go horribly awry, but I did this to make the difference even more obvious. Here is a field of embryonic cells in a zebrafish imaged without phase optics — we’re just seeing the subtle shifts in intensity of the light as it passes through. (If you’re interested, you’re seeing the margin of the expanding epiblast in a pre-gastrula embryo, with the yolk syncytial layer at the bottom of the field.)

Now what I’ve done is slipped in the various prisms and polarizers for DIC, and jacked the contrast up to 11. It’s ugly, but you should be able to see the vivid difference you can get by playing with phase; in particular, notice the great big YSL nuclei at the bottom right. The point here is that we can visualize all kinds of transparent objects in the embryo just by adjusting the widgets on the scope.

There are drawbacks. For one, all that glass diminishes the light intensity at every step — you need a good bright light source to start with. It takes some finesse to use properly (see photo above), and increases the complexity of configuring the scope by quite a bit. I’ve tried to train many students to use this stuff, and most of them throw up their hands in horror and get completely confused — too many knobs! Too finicky! It requires esthetic judgment! Good grief, it’s like…art or something.
This is the usual state I find my microscope in. Those of you who know what I’m talking about will see instantly that both the Wollaston prism and analyzer sliders have been pulled out of the light path. I usually feel like I’ve done well if I teach them to adjust the condenser properly. Grumpy aside: have you noticed how often student scopes have no adjustable condenser at all? You don’t know how to use a microscope if you don’t know how to set up Köhler illumination.
One last picture to show you that DIC can look very pretty. This is the tail bud of a 21 hour embryo; you can see somites with myoblasts, the notochord, mesenchyme, all kinds of cool stuff if you know what you’re looking at. And you can see it all in a transparent embryo.


Whoa. Whoa. Slow down. “Light…is a…wave.”
Is this going to be on the test?
if light is a wave, then explain the photoelectric effect. photons go in, photons go out, never a miscommunication.
Wouldn’t building an fMRI scanner on a Pharyngula scale solve all your problems? The thingy shouldn’t cost much, I guess. (Copyright reserved.)
Not advertising but have you heard of the Lytro camera? Uses light field sensing and photographs contain focus data (focus can be changed after image capture) and paralax info. Might something like that be useful?
@robb
#2
Even better, try explaining the double slit experiment. Especially one involving interference pattern using just 1 photon at a time.
How much of science work is about the subject of research, and how much is about the $#&% instruments between the researcher and the subject?
You say “polarity”, but what you really mean is “polarization”, which is the orientation of the light wave’s electric field.
Darn biologists trying to explain physics!
I see you have a Leica scope. Is this an anti-Kw*k measure?
And I thought it was annoying that my H-alpha filter isn’t exactly parafocal with my other filters…
As a researcher in x-ray phase contrast imaging, I just wanted to say well done & thanks – great to see the optics getting its place in the lime light.
robb, #2 (and anyone else interested):
The photoelectric effect says more about the absorption process than about the nature of light: as Einstein pointed out, beer comes in bottles, but that doesn’t tell us much about beer. More important for demonstrating the photon concept is the auto-correlation experiment, and experiment I have done, whereby light from a single (artificial) atom is divided at a beamsplitter to two different detectors and shown to never produce coincident detection events. When I did this, I used a microscope with about 3 m between objective and ‘eyepiece’ (rather different to PZ’s puny toy, above). The single photons whizzed across my optical table, one at a time (separated by 13 ns), and I never even needed to turn off the lights in the lab! Related to the above comment, though, this proof of single-photon-ness required the light to be filtered by a monochromator – a process relying totally on the wave-nature!
Thanks for this awesome review in optics and how the field applies to biology!
“Light…is a…wave.”
It would be more proper to state that light has “wave-like” properties, not that it is a wave. Technically, QED treats light as particles with phases that take all possible paths between the source and destination. In other words, in the two slit experiment a single photon takes every possible path from source to destination and self-interferes. “Waves” cannot explain the subtle actions that QED explains very well (QED is literally the most numerically accurate theory in all of physics, beyond 10 decimal digits of accuracy).
“Notice how in the previous picture of the amplitude, the peaks in one line up with the peaks in the other, and the troughs line up with the troughs?”
For less confusion among the non-physicists you should have stated “… previous picture of the [same wavelength] amplitude[s]”, since the previous picture compared two different wavelengths.
I also agree with Asad that you should change “polarity” to be “polarization” since that is clearly what would be a correct description. Changing “polarity” merely changes peaks to troughs and visa versa.
With regard to teaching students DIC microscopy, I would recommend introducing them to polarized microscopy first (with lab work) and then proceeding to DIC. I have a very well equipped Zetopan Large Research Microscope with Polarization and DIC among *many* other options, and I self-learned polarization (using a Walter McCrone book) followed by DIC, and it was really quite easy this way.
Sorry about the formatting, I don’t know how to retain my original format in the posted version.
Yeah, P. Z. is wrong when he makes that claim that light is a wave.
However, it seems to me that this was an intentional simplification in an effort to provide a simple explanation of certain features of Newtonian optics and that going into the more subtle details of advanced physics was not appropriate for that task.
Oh no! He erased the board before I finished writing everything down! Curse my thumbless paws.
This reminded me of a Larry Niven short story, “For a Foggy Night,” in which someone explains that fog is really an encroaching alternative-universe portal. It’s not caused by water in the air; water is transparent, after all.
Oh, yes, an in addition to
War is Peace
Freedom is Slavery
Ignorance is Strength
remember that
Time is Space
Matter is Energy
Particles are Waves.
Excellent article, PZ. All I knew about optics before was Herschel wedges, pentaprisms, diffraction gratings, left-hand and right-hand sugar, and varifocal lenses. ;-)
<physicist>*twitch*</physicist>
“Light is a wave”…”light is a particle”…
I prefer “light is one of the ways the universe reminds us not to anthropomorphize how it thinks things should work.” Or just “light is weird as hell, but in the awesome way.”
This is great. Thanks, PZ!
It’s always best if someone actually understands in detail how his tools function. (You’d weep if I told you the number of Silicon Valley engineers to which I’ve had to explain Ohm’s Law or similar basic concepts).
Hmmm, wonder if we could modify the 2nd Amendment to require the ability to explain how gunpowder pushes a bullet out a gun barrel (including demonstrating a knowledge of redox reactions and the gas laws) before anyone is allowed to purchase a gun?
What a lovely thing to read. :)
Being shown and taught DIC was one of the first experiences during which I really fell in love with biology. It’s all just so beautiful.
QED Q.E.D.
Re: fMRI, MRI doesn’t have great spatial resolution. It’s a popular medical imaging technique because it has excellent contrast between nearby tissues (things with similar density don’t necessarily have similar chemical properties), but most medical scanners cobble images together from voxels on the order of 0.5 mm x 0.5 mm x 1-3mm, so they wouldn’t be super useful to follow development on a cellular level.
I received a microscope for my birthday. It has a camera that can be hooked up to a computer and a device for preparing permanent slides, much of which intimidating to one who never had to do any of that at school or college and is much more into the theoretical side of things. Still, I was proud of it, but now I feel quite emasculated. Damn. The professional biologist’s microscope is bigger and better than mine. I should have expected this. Why didn’t I expect this?
I sense a disturbance in the electromagnetic force.
This thread is quiet … too quiet.
Naked Bunny, don’t listen to those biologist types … physicists know light is a particle. At least some of the time. Yeah, it will be on the test, so don’t forget to bring your colored pencils.
Actually, PZ, that was a pretty good explanation. Cool bits of hardware FTW. For all the various optical equipment I’ve touched in my life I’ve never come close to a microscope of any decent quality.
Callinectes – have you ever heard the saying about cameras “the best camera is the one you have with you”? The best microscope is the one you have and use. I’ve used all kinds of microscopes, from 60 year old student dissecting scopes with mirrors instead of lights to electron microscopes, and I’ve never had one at home that I could use any time I want, much less be able to easily capture images digitally.
By the way, what is the device to prepare slides? I can’t think of what that might be, unless it’s some sort of mini-microtome for slicing.
Cautious qualification noted. They can, in some circumstances, and the effect is Haidinger’s brush. And for an added bonus there’s a cephalopod page!
Both QED and (more recently) relativity have reached 18 digits.
“And don’t anthropomorphize nature, she hates that.”
I’ve taken useful photos of fossils by just holding a digital camera to one ocular of a binocular microscope.
Also, QeD is Klingon for “science”.
So, you’ve just been DIC’ing around with your microscope?
…sorry.
Perhaps to satisfy the physicists out there, PZ could have takes a side trip and mentioned that light, for the purposes of this essay, can be very adequately modeled as a wave. If instead this was an essay not on microscopy, but on what happens when you play with half-silvered mirrors and multiple paths or send single photons through double slits, in that case it often makes a lot more sense to think of light as little particles. But we’re not talking about the weird things that happen with double slits. This is about fishies.
While the explantion on microsopy is educational, ‘rubes’ is short for Reubens which is a an ethnic slur. Likely an ‘honest oops’ here. As the saying goes: the more you know.